- | Regulation Regulation
- | Data Visualizations Data Visualizations
- |
FDA Regulations Should Be Safe and Effective
Before approving medical drugs sold on the market, the Food and Drug Administration (FDA) requires companies to demonstrate that the drug meets basic standards of safety and effectiveness. This is a high bar to pass, and Americans expect that officials at the FDA will use sound judgment in deciding which drugs get approved, since these decisions will have profound effects on the health and well-being of the American people.
Before approving medical drugs sold on the market, the Food and Drug Administration (FDA) requires companies to demonstrate that the drug meets basic standards of safety and effectiveness. This is a high bar to pass, and Americans expect that officials at the FDA will use sound judgment in deciding which drugs get approved, since these decisions will have profound effects on the health and well-being of the American people. Unfortunately, the experts at the FDA craft regulations based on analysis that would not meet the same rigorous standards the FDA requires from corporations seeking drug approvals. Maybe it’s time that the safety and effectiveness standard applied to drugs should also be applied to regulations.
Are FDA Regulations Safe?
First and foremost, Americans expect regulations addressing risks to make them safer. Unfortunately, regulations designed to reduce risks in one area can sometimes increase risks elsewhere. For example, when the FDA considers approval of drugs to combat life-threatening diseases like AIDS, it has to consider not just the side-effects of these drugs but the potential risks that occur if the agency does not allow the drugs to come to market. These risks include the deaths of patients whose lives could be extended by those very same drugs. Consideration of these so-called risk-risk tradeoffs should be a part of any well-done regulatory analysis. However, agencies only rarely consider risk-risk tradeoffs, despite guidance from the Office of Management and Budget explicitly recommending they do so.
Another example of a risk-risk tradeoff at the FDA arose surrounding health risks associated with consuming methyl mercury, which is associated with some birth defects. In 2004, the FDA and EPA published a joint “consumption advisory” explaining the risks to pregnant women and young children of ingesting mercury through fish. However, fish intake also provides beneficial Omega-3 fatty acids. Had the FDA considered risk-risk tradeoffs more carefully, the agency might never have issued the consumption advisory document, which likely led some women to eat less fish that, on balance, would be good for them and a newborn. In 2008, the FDA considered reversing this policy. However, the old recommendations currently remain on the agency’s website, leaving the public more confused than ever about the risks associated with consuming fish.
Are FDA Regulations Effective?
While estimating the effectiveness of rules is very difficult, especially before they are implemented, one helpful device might be to look at the economic analysis that agencies conduct alongside their most important rules. Since 1981, presidents have required agencies to conduct Regulatory Impact Analysis (RIA) for regulations that have large effects on the economy. The Mercatus Center’s Regulatory Report Card assesses the quality and use of agency RIAs for economically significant proposed regulations. Researchers evaluate the analysis accompanying each regulation and assign a score ranging from zero to 30 points. The average score at the FDA for seven regulations scored since 2008 is 12.6, or 42 percent. One consistent shortcoming is that the FDA often fails to present credible evidence to back up its claims that a significant problem requiring intervention exists in the marketplace. Similarly, the FDA often fails to provide evidence that the regulation the agency is proposing is likely to produce actual benefits to the public.
The charts below demonstrate this in more detail by showing scores for two criteria from the Regulatory Report Card. Criterion 1 asks how well the agency identified and proved the existence of a market failure, government failure, or other systemic problem a regulation is supposed to solve. As the chart shows, the FDA is often able to name a systemic problem a regulation is designed to address. However, when asked to outline a coherent theory explaining what caused the problem, support this claim with empirical evidence, or analyze the inherent uncertainty surrounding the issue, the agency does more poorly.
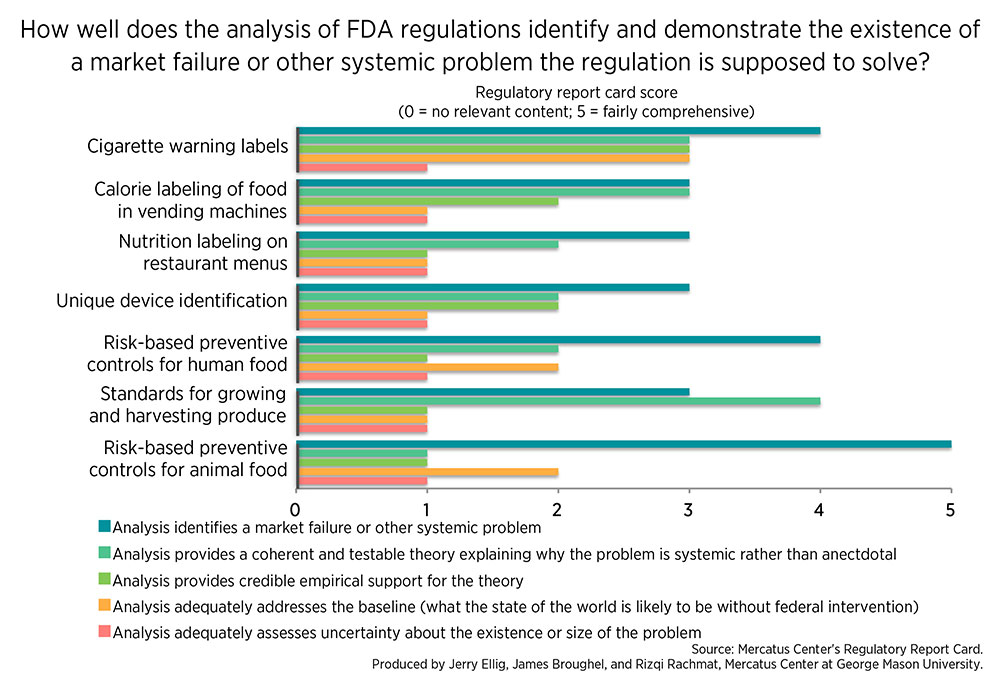
Criterion 3 of the Report Card asks how well the agency analyzed the benefits or other desired outcome the regulation is designed to address. Again, we see the same general pattern: the FDA can name a desired outcome in many cases, but it often fails to provide a coherent theory about how the regulation will achieve the outcome or empirical evidence that the regulation is likely to achieve its goals.

A recent proposed regulation requiring Risk-Based Preventive Controls for Food for Animals illustrates both types of shortcomings in FDA regulatory impact analysis. The FDA claimed animal food safety is suboptimal because consumers and purchasers lack sufficient information on the safety attributes of foods and the sources of food contamination. Yet the RIA contained no information about the actual state of customer information, the very market failure the FDA claimed it was addressing. The RIA merely presented some statistics on animal food hazards and recalls without any contextual information that showed just how large the problem was.
The analysis of benefits shared similar shortcomings. The RIA claimed reduced risk to animals and humans handling animal food, as well as to humans who eat meat or animal products, as benefits of the regulation. Yet the RIA just asserted that contamination would be less likely in the presence of the regulation without backing up the claim with any empirical evidence. The RIA also claimed that the regulation would reduce the number of costly recalls, despite evidence that similar regulations for human food have increased the number of recalls.
These analytical shortcomings are very troubling because the FDA added more than 1,000 regulatory restrictions to the Code of Federal Regulations between 2008 and 2012. Regulatory restrictions are words that signify a binding constraint that restricts behavior. These are words like “must,” “shall,” or “may not” that appear in regulatory text.
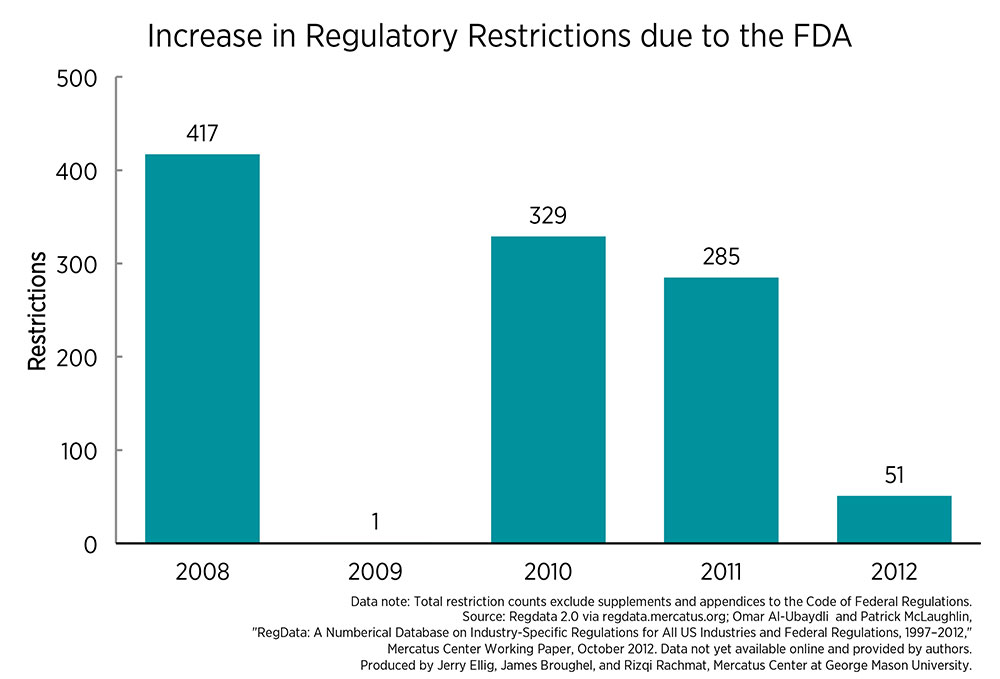
At the same time, Congress has steadily increased the FDA’s funding. FDA spending increased 65 percent in real terms between 2008 and 2012 and by more than 270 percent between 1990 and 2012. Given these large increases, one has to wonder why the FDA is not producing better analysis to support its proposed regulations.

The FDA Standard Applied to Itself
If medicine must be shown to be both safe and effective before it is sold on the US market, why not hold regulations to the same standard? To improve the safety and effectiveness of FDA regulations, Congress could mandate that the FDA meet basic standards for regulatory analysis. This should include a requirement that the FDA consider risk-risk tradeoffs, demonstrate the existence and cause of a significant problem the agency is trying to solve by regulating, and provide evidence that the regulation will actually achieve the desired outcomes. This way, the agency can be held accountable if the regulation does not achieve its desired goals. Safety and effectiveness are high bars that regulated entities are asked to meet. Why should the American public expect anything less from the FDA itself?
1 Economically significant” refers to regulations having an annual impact of $100 million or more on the US economy.
2 Average scores on the last four questions in Criterion 1 are all statistically different from the first question.
3 In this case, average scores on the first two questions, which ask whether the agency named and measures the outcomes, are not significantly different from one another. Scores on the remaining questions are statistically significantly different from the first question.

